We want to hear from you! Take the survey.
How do you use It’s Your Yale? How can it be improved? Answer for a chance to win Yale swag.
News (Archive)
February 7, 2023
New Yale HRPP Policy and Standard Operating Procedure Manual, Investigator Manual, and Additional Documents
The Yale Human Research Protection Program (HRPP) is excited to announce the launch of the new Yale HRPP Policy and Standard Operating Procedure Manual, with an effective date of January 30, 2023. This purpose of this manual is to consolidate existing standalone Yale HRPP policies, procedures, and select guidance documents into one streamlined manual. The manual also includes new topics related to human subjects research conduct and oversight, reflecting the most up-to-date regulatory information. We are confident our Yale research community will find this new manual both valuable and easy to navigate.
The Yale HRPP has also updated: 1) the HRPP Investigator Manual, which provides detailed information on research preparation, IRB submission, and study team responsibilities, and 2) checklists, worksheets, submission forms, and templates that aid in successful application submission to the Yale IRB.
Training sessions on these updated documents were provided to the Yale research community throughout January 2023. Please review the session slides and a high-level summary document that outlines the purpose and major points pertaining to each new/updated document below. All referenced documents can be found on the Yale HRPP website. For any questions, please contact HRPP@yale.edu.
New Yale HRPP/IRB Manuals & Related Documents
Summary of Yale HRPP Document Changes
Recording: Yale Research Community Training January 2023
October 11, 2022
Magnetic Resonance Research Center Protocol Committee Review (MRRC PRC) ancillary review process
Beginning October 11, 2022, the process will be changing for obtaining MRRC approval for research using the MRRC facility. Investigators will no longer request review of their research by directly contacting the MRRC via email. Instead, the HRPP Institutional Review Team (HRPP-IRT) will submit the request to the MRRC on behalf of the Investigators. This will occur when there is a new protocol that makes use of the MRRC or when there is a protocol modification that involves the MRRC.
What this Means to You
Investigators should visit the MRRC website for information on what the MRRC PRC will be looking for in the consent forms and protocol.
Investigators will still be required to fill out the Proposal for use of MRRC Resources form found on both the MRRC website and in the IRES IRB library. This form is to be uploaded into IRES IRB under Local Site Documents/Other Attachments as part of your submission to the IRB.
The HRPP-IRT will conduct the ancillary review process to obtain MRRC PRC approval, before sending the protocol to the IRB for review. If the MRRC PRC requests revisions the IRB will inform the investigator via IRES IRB. Once the investigator submits the requested revisions back, the HRPP-IRT will request review of the revisions by the MRRC PRC.
When final MRRC PRC approval is obtained, the HRPP-IRT will receive documentation of the approval for our records, the PI will receive communication via email from the MRRC PRC and if all other IRT revisions are complete the protocol will be sent to the IRB For review.
July 12, 2022
Changes regarding Yale IRB and HRPP Review Fees
On July 12, 2022, Researchers, Research Staff, and Business Offices received emailed communication from Pamela Caudill, Senior Associate Provost, Research Administration & Institutional Official; Brian Smith, Deputy Dean for Research, (Clinical and Translational); Co-Director, Yale Center for Clinical Investigation (YCCI); Arnim Dontes, Deputy Dean, YSM Finance & Administration, and Linda Coleman, Director, Human Research Protection Program regarding upcoming changes in Yale IRB and HRPP Review Fees.
The communication explained that effective September 1, 2022, Yale University is implementing a change to the process regarding IRB and HRPP review fees. There is NO change to the HRPP/IRB fee schedule or which studies are considered billable.
Detailed description of the change:
- Currently, the HRPP invoices IRB and HRPP fees in two ways: 1) by charging the sponsor directly (YCCI has done it on the HRPP’s behalf), or 2) by creating journal entries in Workday for certain billable activities.
- Effective September 1, 2022, Yale is streamlining this process so that ALL billable activities are charged by the HRPP to the departments via journal entries. YCCI will be available to help to recoup the money from the sponsor on the department’s behalf; however, the financial responsibility rests solely with the Department.
- Note: If a department has NOT used YCCI services for sponsor invoicing, it can either recoup the money from the sponsor on its own or it can consider using YCCI to help collect it on its behalf. If the investigator would like to have YCCI start working with the investigator’s team to recoup money from the sponsor on their behalf, the investigator’s Business Office can contact YCCI Billing at irb.invoicing@yale.edu.
How the change affects investigators:
- The HRPP will charge for IRB review at the time of a final IRB approval/determination, beginning with approvals/determinations issued on or after September 1, 2022. The HRPP will ask for the Chart of Accounts (COA) number, the name of the Business Office contact, and other associated information in the IRB/HRPP Submission Form. Yale understands that at the time of initial IRB review, the sponsor award account is not yet available. Business Offices are asked to have a general departmental COA available so that the initial review fees can be charged to this account. Alternatively, if an “at-risk” account has been created, then that COA may be supplied. All following submissions will be charged to the COA for the award (or the general COA until the COA is set up).
- When an initial study is submitted for IRB review or a request is made to use an external IRB, the HRPP Institutional Review Team designee will notify the Business Office contact via email that a new invoiceable submission has been received. This will alert them to expect a journal entry when a final IRB approval/determination or an authorization to use an external IRB is issued.
Available training and follow-up information
- A live virtual information session regarding IRB and HRPP billing is scheduled on August 16, 2022, at 9 am. Registration is available through Training Management System.
- The HRPP will also post a recorded slide presentation on the HRPP website after this session.
Questions regarding this notice should be directed to hrpp@yale.edu or irb.invoicing@yale.edu.
August 19, 2021
Research Activation
Effective August 16, 2021, safety protocols related to conducting human subjects research during the pandemic, will be reviewed by the Human Research Protection Program (HRPP) before the IRB review. In certain circumstances, the HRPP will consult with the representatives from the Office of Environmental Health and Safety, Off Campus Research and Fieldwork Committee, and Human Subjects Research Review Committee, who have conducted these reviews since the beginning of the research reactivation phase.
Investigators will submit safety plans in IRES IRB, the HRPP electronic system. EHS Integrator will no longer be used for this purpose.
Instructions
- Investigator must complete ‘Supplement to IRB Submission and Requests to Use External IRB – Safety During Pandemic’ form available in Other Forms tab in IRES IRB Library. Exception: investigators conducting secondary data analysis research and clinicians requesting an emergency use of a test article or device do not need to complete this form.
- The form should be uploaded as a supporting document along with the research documentation and submitted in IRES IRB.
- The HRPP will contact the investigator and primary contact for the study within 2 business days should there be any questions about the proposed safety plan.
- Once the safety plan is accepted, the study will proceed to the IRB for review or will receive authorization to use external IRB.
August 7, 2021
IRES IRB Upgrade
The IRB and HRPP electronic system, IRES IRB, is scheduled to undergo an upgrade to the most recent version of the system. The upgrade is scheduled to begin after 5 pm on Thursday, August 12th. It should become available to users on Tuesday, August 17th, mid-afternoon hours. There will be no access to IRES IRB between these dates.
There is no change in the schedule of the IRB meetings. In case of emergencies and urgent actions that require IRB approval, contact the HRPP office at hrpp@yale.edu. The IRB will be able to conduct urgent reviews during the time the system is down.
What changes to expect
There are three types of changes that are being implemented: changes to navigation, changes in questions on pages in the electronic submission, and changes to the submission and reviews processes. Examples of some of these changes are listed below:
- Navigation
- My Inbox moves to a tab called Dashboard
- Shortcuts to Library, Help Center, IRB Submissions, IRB Reports move from Menu on the left side to the top of the screen
- The study record shows a MENU of all pages on the left side of the screen to visually represent progress while completing the electronic application
- Overall feel and look of the system is changed to allow for an easier navigation
- Screens
- Questions are removed from several pages of the electronic application
- A new page with research location is added
- New questions regarding repositories and sharing of the specimens are added
- Supporting Documents page is consolidated with local consent and recruitment materials page
- Processes
- Changes are introduced to the process for requesting use of external IRBs and updating the system after the study is approved by the external IRB
- Several minor changes are made to the internal IRB processes with no impact to the researchers
First Actions after the Upgrade
The upgraded system will include new questions on two existing study screens (Study Scope and Use of Technology in Research) and a new page (Local Research Locations). The estimated time to answer these questions is less than 5 minutes. Investigators will need to provide answers to the new questions and verify information on the new page at the time of the first modification on the protocol or before any pending modification or initial study receives IRB approval. A new function called ‘Validate’ is available to help identify missing answers throughout the electronic application. See below for details:
First Modification, Continuing Review with Modification, Study Update on an approved protocol
Before submitting these actions to the IRB, go through all screens to verify and correct the information on all pages. Answer any missing questions.
First Continuing Review on an approved protocol
No changes can be made to the protocol record at the time of continuing review. If you wish to provide answers to the newly added questions, you will need to create Continuing Review and Modification. Pending Continuing Reviews will be processed as usual.
Modifications, Continuing Review with Modifications, and Initial Submissions pending IRB review
You will be asked by the IRB and HRPP staff to provide answers to the newly added questions before the IRB approval is issued. If the submission receives a determination of Modifications Required to Secure Approval or Deferral, you will need to answer the new questions before submitting your response in IRES IRB.
Updates to studies under external IRB purview
Any pending update to the study under an external IRB purview will be discarded at the time of the upgrade. Updates currently pending HRPP acknowledgment that cannot be successfully processed prior to the upgrade (as a result of a pending ancillary review) will be recreated on behalf of the PI after the upgrade.
Training
Recorded sessions on changes in the new system and revised quick guides will be available on the HRPP website prior to the upgrade. They will also be posted in the Help Center in IRES IRB once the upgrade is completed.
There are two different types of live training sessions available via Zoom throughout the end of August:
- Changes in IRES IRB for studies under Yale IRB purview
- Changes in IRES IRB for studies under External IRB purview
Registration is available through Training Management System.
Sessions on using IRES IRB for individuals who have not used the system before will resume in September.
May 1, 2021
Procedure on Research Staff Engaged in Research Projects
Effective May 1, 2021 a new procedure was published regarding listing research staff members in the IRES IRB record
The research record in the IRB electronic system, IRES IRB, must include the names of at least the following team members engaged in human subjects research under Yale IRB or HRPP purview:
- Principal Investigator (PI),
- PI Proxy, if one is identified for the study,
- Investigators,
- Individuals external to Yale who require a reliance agreement OR Unaffiliated Investigator Agreement, and
- Other members of the research team who report financial interests related to the research.
Team members who do not meet these criteria do not need to be listed in IRES IRB.
If the Principal Investigator wishes to list all of the members of the research team in IRES IRB, the study team members page in the system includes a selection of available roles to further describe research roles of each individual.
Note: The IRB can always require listing additional members of the research team.
September 1, 2020
September Updates
Effective September 1, 2020, the following changes are being implemented to the HRPP and IRB practices:
- The Yale IRB increase number of meeting from a minimum of 10 times per month to meeting a minimum of 20 times per month by adding new IRB panels;
- New submission deadlines are being introduced;
- Watermarking of documents practice is revised so that the approval watermark will no longer be applied at the time of IRB approval of the continuing review;
- The dates for the second and all subsequent continuing reviews of research will be based on the date the continuing review is approved (with or without modifications) by the IRB.
View the detailed announcement that was sent to the research community on September 1, 2020.
July 7, 2020
Release of New IRB and HRPP Documents
This notice describes changes to the existing IRB documents and changes that have been implemented in July 2020 to the HRPP and IRB processes. Please contact the hrpp@yale.edu with any questions regarding the changes and how it will affect your research.
Summary
July releases:
- New to Yale IRB
- New Protocol Templates and access to a Protocol Builder too
- Revised Consent Glossary
- Parental Permission and Assent Forms
- Certificate of Confidentiality Quick Guide
Detailed Description
New Yale IRB Submission Form
Change
The previous version of the submission form, called Yale Site Addendum, is no longer be available in IRES IRB for use as of Monday, July 12, 2020. A revised submission form will be posted in IRES IRB. The new document consists of required sections that must be completed for all submissions of non-exempt research to Yale IRB and supplemental sections that are required only for certain types of research. The form includes extensive guidance and walks investigators through some of the more complicated regulatory requirements (e.g. FDA regulated research with devices, etc.). A recorded training will be posted later in the summer.
What this means for your research
The new Submission Form must be used for all new non-exempt research regardless of funding and uploaded in the Supporting Documents page in IRES IRB. Research already approved is considered grandfathered-in and does not require researchers to submit the new form.
If you already started completing the existing Yale Site Addendum for an initial submission that has not been sent for IRB review, you can still use that document for the next 3 months. However, Yale IRB will stop accepting initial submissions utilizing the outdated form on October 19, 2020.
New Protocol Templates and Protocol Builder access
Change
A well though-out and well-written research protocol leads to better quality research and makes the IRB review processes more efficient. To help Yale investigators develop quality investigator-initiated research protocols, the following is available to all investigators:
New word versions of protocol templates in IRES IRB for researchers to use without use of any additional software:
- Research with Devices
- Research with Drugs
- Observational Study of Individual or Group
- Repository
- Social-Behavioral Research
A new Protocol Builder tool to help develop protocols. (The use of this tool is not required.)
- YCCI and HRPP acquired a license to Protocol Builder, a web-based application that offers a wide range of protocol templates that can be utilized by researchers conducting biomedical and social, behavioral, and education research studies. Protocol Builder can be assessed by any member of Yale with an active Yale netID at https://app.protocolbuilderpro.com/register/yale-university. Once you complete a protocol in Protocol Builder, you can generate a WORD or PDF file of the document and upload the document into the IRES IRB system.
Other publicly available templates such as the following:
- National Institutes of Health (NIH) e-Protocol Writing Tool or word versions of the NIH templates for Phase 2 or 3 clinical trials that require Investigational New Drug applications (IND) or Investigational Device Exemption (IDE) applications and Behavioral and social sciences research; and
- Protocol templates available via TransCelerate suite. You can learn more about the Clinical Content and Reuse Initiative, which made the sharing of common protocol template and statistical analysis plans possible, by watching a video created by TransCelerate.
Yale does not require a use of a specific protocol format as long as all of the elements of a research protocol are included in the document.
What this means for your research
The Yale HRPP and IRB will continue accepting protocols authored by external entities such as cooperative groups or industry sponsors. In addition, previously approved IRB approved research does not need to be transferred to the new Protocol Template.
The previous versions of the non-exempt biomedical and social behavioral research protocol forms are no longer be available on IRES IRB for use as of July 12, 2020. Because we recognize that it is a significant change for researchers and research coordinators you may continue to use an older version of the protocol application (and submission form) for 3 months. Yale IRB will stop accepting initial submissions utilizing the outdated templates on October 19, 2020. Again, previously approved studies are considered grandfathered-in and no changes are necessary.
Training
Protocol Builder offers instructional videos and training that can accessed when you log in. A webinar for Yale users will be offered early in the fall. It will be recorded and posted on the Protocol Builder page. You will be notified about the exact date.
Where you can direct your questions
If you have questions about this new resource, you can contact HRPP Office at hrpp@yale.edu. If you have technical questions about how to use Protocol Builder, please contact their customer support at 844-563-1042.
Revised Consent Glossary
A revised Consent Glossary with new suggested language and numbering is available in the Consent Forms tab in the Library section of IRES IRB. Refer to the glossary when drafting consent forms for human subjects research.
Parental Permission and Adolescent Assent Forms
The Parental Permission form was revised to meet the regulatory standards of 2018 regulations. The Adolescent Assent form was also revised to offer simplified language accessible to adolescent participants in research studies. No changes are required for ongoing, IRB approved research. Both templates are available in the Consent Forms tab in the Library section of IRES IRB.
Certificate of Confidentiality Quick Guide
As a result of recent changes to the process of applying for a Certificate of Confidentiality from NIH for non-NIH funded studies, the HRPP created a quick guide on the topic. It provides researchers with information needed to submit a request for a Certificate of Confidentiality. It is available in the Handbooks and Manuals tab of the IRES IRB Library.
If you have any questions about this notification, please, contact Yale HRPP.
April 28, 2020
Yale COVID-19 Human Subjects Research Oversight Committee Process
Yale University is committed to conducting research on COVID-19 to combat the ongoing Pandemic. To streamline and fast track study activation and implementation, Yale established a COVID-19 Human Subjects Research Oversight Committee process. The announcement about the process was sent to the research community. Questions should be sent to Yale Clinical Research Resources team at clinicalresearchresources@yale.edu. Additional information is available on the Yale COVID-19 page (“Research, Clinical & Data Driven Responses to COVID-19”)
December 2, 2019
A new requirement for review of research protocols with ionizing radiation by the Yale University Radioactive Investigational Drug Committee (RIDC) and a revised process of submission of protocols for Radioactive Drug Research Committee (RDRC) review.
Background
Research with unapproved drugs or biologics require an FDA approved IND for the drug/biologic to be used in research. Per 21 CFR Part 316 certain studies that use the radioactive materials can be conducted without an IND if Radioactive Drug Research Committee (RDRC) approves the study in lieu of the FDA. The criteria for when RDRC review is allowed is listed at the bottom of this communication.
Studies with an FDA approved IND, FDA-approved radiopharmaceuticals, or FDA allowed IND exemption do not undergo the review by RDRC. For years, Yale investigators conducting research with such agents submitted their protocols to Yale New Haven Hospital Radiation Safety Committee to evaluate radiation dosimetry even though the research was taking place at Yale PET Center and not the hospital. To close the gap and provide the investigators with the necessary review at Yale, the Yale Office of the Provost established Radioactive Investigational Drug Committee (RIDC). Dr. Albert Sinusas agreed to serve as the Chair of the Committee. The HRPP will help with administrative support. Studies reviewed by the RIDC will no longer require a review by Yale New Haven Radiation Safety Committee unless research related radiation is also used in hospital.
Role and Responsibility of the Radioactive Investigational Drug Committee
HRPP Guidance 940, ‘Guidance on Committee Reviews Required for Human Subjects Research Protocols Using Radiation’, describes roles and responsibilities of each Committees that may review the research with radiation.
The following is the summary of the RIDC responsibilities:
- Ensures the underlying science and research protocol are of sound design.
- Reviews and approves the consent form language related to the radiation.
- Evaluates radiation dosimetry.
- Confirms consistency between the FDA application and Yale University study-related documents.
- Ensures that the PI and research staff are current on any required training.
How it affects your research
If your study is already approved by an IRB, initial review by the RIDC will NOT be required. Any modifications to your protocol that affect the use of radioactive materials will require RIDC review. The HRPP is creating a database of all the existing studies that will fall under RIDC purview. Basic information about your study will be provided to RIDC. HRPP will work in collaboration with PET Center to obtain the necessary information if the IND is held by the PET Center. The RIDC review does not replace the requirement for Yale Radiation Safety Committee review.
Submission Process for Initial RDRC/RIDC Review or Modification
Protocols requiring RIDC or RDRC review will be sent to RIDC/RDRC using IRES IRB. The PI’s responsibility is to fill out an Application to Involve Human Subjects in Biomedical Research with Ionizing Radiation and RSC Human Use Review Cover and upload both as Supporting Documents. When studies are submitted in IRES IRB, the HRPP staff will request review from both RIDC/RDRC and Yale RSC, and electronically notify PET Center about the upcoming research protocol. RIDC/RDRC and RSC will review the protocol and submit their findings in IRES IRB.
The workflow and details are below:
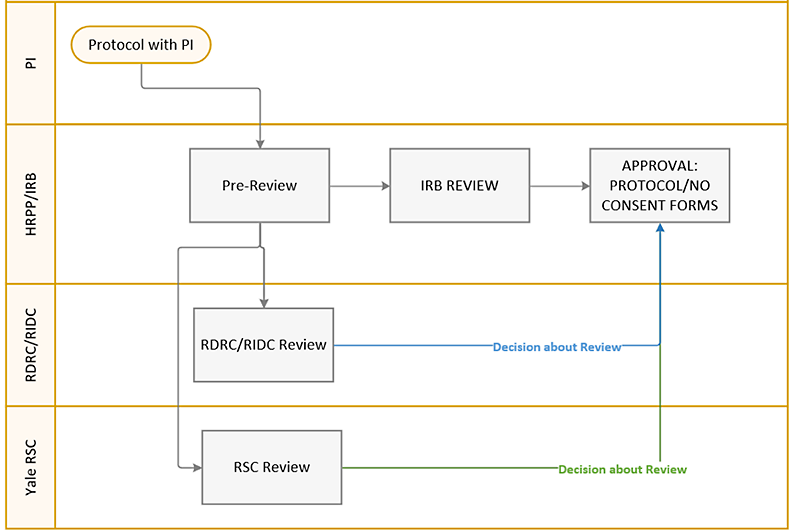
Upon submission of the protocol in IRES IRB, the HRPP staff will:
- Verify that the RDRC/RIDC Request Form is completed and submitted in IRES IRB,
- Verify that the RSC Human Use Review Cover sheet is uploaded in IRES IRB,
- Confirm that the PET Center required language is included in the consent forms (see Consent Glossary document in IRES IRB for the language),
- Confirm that the documents are uploaded appropriately,
- Verify that the Authorized User is listed on the protocol,
- Notify RDRC/RIDC, RSC, and PET Center contacts via IRES IRB about the protocol submission.
Upon receipt of the notification that a protocol requires RIDC/RDRC review, the committee will schedule the protocol to the next available meeting, review it, issue determination, and upload the determination in IRES IRB:
- Approval with no comments to the IRB,
- Approval with recommendations/comments to the IRB,
- Revisions Requiring RIDC/RDRC rereview,
- Conditional Approval/Revisions Needed NOT Requiring RIDC /RDRC Rereview.
Upon receipt of the notification that a protocol requires Yale RSC review, RSC will review the submission, issue determination, and upload the determination in IRES IRB:
- Approval with no comments to the IRB,
- Approval with recommendations/comments to the IRB,
- Revisions Requiring Yale RSC rereview,
- Conditional Approval/Revisions Needed NOT Requiring Yale RSC rereview.
IRB Actions:
- IF RIDC/RDRC/Yale RSC Approved: Modification is submitted on behalf of the PI to release consent forms.
- If Revisions Requiring RIDC/RIDC/RSC Re-review: Modification is created and submitted in IRES IRB on behalf of the PI by the HRPP staff and the RIDC/Yale RSC decision letter is sent with any required modifications. The workflow is repeated but IRB approval NOT issued until approval is obtained.
- If Conditional Approval/ Revisions NOT Requiring Re-review: Modification is created and submitted in IRES IRB by HRPP staff on behalf of the PI and the RIDC/RDRC/Yale RSC decision letter is sent with any required modification.
Contacts
If you have questions about the RIDC or RDRC review process, please contact the following individuals:
- Amy Blakeslee, HRPP Manager at amy.blakeslee@yale.edu – Amy is the liaison between the Yale HRPP and the Yale IRB. She is available to help the investigators fill out the RIDC form and help calculate the dosimetry. She will work closely with PET Center to ensure the information you provide is accurate.
- Donna McMahon, RIDC Manager, at ridc@yale.edu – Donna provides administrative support to RIDC. She will submit the RIDC determinations in IRES IRB.
Update Regarding Revised Consent Forms, Consent Glossary, and Instructions for Converting Studies to Revised Common Rule
June 28, 2019
Changes to HRPP and Yale IRB Fees for Commercially- Supported Studies
The announcement regarding the HRPP and IRB review fees was sent to the research community. Please, contact hrpp@yale.edu with any questions.
January 22, 2019
The purpose of this communication is to provide you with updates since our last communication earlier in the month regarding the implementation of the revised regulations governing research involving human subjects, the “Revised Common Rule”, which went into effect yesterday, January 21, 2019.
This communication addresses the following aspects of the Revised Common Rule that may be important to you:
- Updated Consent Templates
- New Consent Glossary
- Updated Request for Exemption Determination
- Instructions for Continuing Review requests for a study to be converted to Revised Common Rule standards.
1. Updated Informed Consent Templates
Three revised consent templates were uploaded into the Library section of IRES IRB:
- Compound Authorization and Consent for Biomedical Research (HIPAA and Consent)
- Compound Authorization and Consent for Social, Behavioral, and Educational Research (HIPAA and Consent)
- (SBE) and Consent template for SBE research (without HIPAA elements)
The new consent templates include new required elements of consent, revised guidance for drafting of the consent forms, and updated suggested language.
Please, note that the purpose of the consent templates is to help investigators draft their consent documents when no other templates are provided. If you received a template for your research from other sources, either a sponsor of the study or the coordinating center, there is no need to revise that template to follow Yale’s suggested form. You should work with the sponsor to revise the consent for templates as necessary such as to add Yale required language.
In the next few weeks, the HRPP will be posting the remaining templates: parental permission, repository template, and assent forms.
The consent forms will be reviewed and updated based a predefined schedule unless there is a required regulatory/university policy change, significant human subject protection issue, or other reason that necessitates an immediate change.
2. New Consent Glossary
A Consent Glossary contains a compilation of commonly used terms and descriptions of research procedures organized in an alphabetical order. As noted in the Glossary, some of the language may be required (by applicable legal, regulatory, or other requirements such as by the Yale ancillary committees or Yale policy) and some may be preferred (but not required).
Review the terms in the Glossary to select the paragraphs that you may need to use.
The Glossary document will be reviewed and updated based a predefined schedule (e.g. quarterly, biannually, annually, etc.) unless there is a required regulatory/university policy change, significant human subject protection issue, or other reason that necessitates an immediate change.
The Glossary includes references and the reading level of each section. The goal is to use language that is at grade 8 or below, when possible. A few sections (e.g., in case of injury, GINA, etc.) were not revised or minimally edited, but will be reviewed later.
The Glossary is uploaded in the Library Section of IRES IRB.
3. Updated Request for Exemption Determination
A revised Request for Exemption Determination was uploaded into the Library section of IRES IRB. As a reminder, Yale-specific category 7 was removed as the revised rule includes a new exemption category 3 (i) (A), (B), and (C), which allows for certain benign behavioral interventions to be considered exempt. As such, there is no further need for the largely redundant Yale-specific category 7. In addition, due to lack of agency guidance related to requirements and use of broad consent, Yale IRB will generally not grant exemption determinations utilizing categories 7 and 8. As such, they were not included in the Request for Exemption Determination. The Yale HRPP will re-evaluate the use of broad consent after the Department of Health and Human Services (HHS), Office for Human Research Protections (OHRP) issues relevant anticipated guidance.
4. Instructions regarding Continuing Review
- For research submitted to the Yale IRB for approval after January 21, 2019 (without FDA or DOJ oversight), the Yale IRB will approve the following types of studies without requiring Continuing Review unless the IRB determines there are special circumstances that requires ongoing continuing review: (1) applicable minimal risk research; and (2) greater than minimal risk research if the research is closed to enrollment and the remaining research activities are limited to data analysis, including analysis of identifiable private information or identifiable specimens, or accessing follow-up clinical data from procedures that participants would undergo as part of clinical care.
- For research approved by the Yale IRB prior to January 21, 2019 (without FDA or DOJ oversight), the Yale IRB will reapprove a study at its next continuing review without requiring further continuing review by converting the study to the Revised Common rule standard. Those include: (1) applicable minimal risk studies; and (2) greater than minimal risk studies if the research is closed to enrollment and the remaining research activities are limited to data analysis, including analysis of identifiable private information or identifiable specimens, or accessing follow-up clinical data from procedures that participants would undergo as part of clinical care.
In order for the Yale IRB to convert the study to Revised Common Rule standard, the research must meet all of the approval criteria of the Revised Common Rule. This means that if a study meets the approval criteria of the Revised Common Rule and does not have consent forms, then the study will be converted to the Revised Common Rule at the time of your next Continuing Review unless the IRB determines there are special circumstances that required ongoing continuing review. However, if your study meets the approval criteria of the Revised Common Rule and has consent forms, the consent forms will need to be revised to include the new consent elements required under the Revised Common Rule. The list of all the elements is included in the Consent Glossary for your reference.
If you would like the Yale IRB to consider converting a study that includes consent forms to the Revised Common rule standard so that the study no longer requires continuing review, at the time of your next Continuing Review (with a CR date of January 21, 2019 or later), submit a Continuing Review with a Modification to upload the revised consent form that includes the following elements:
1. Key Summary (required): Informed consent must begin with a concise and focused presentation of the key information that is most likely to assist a prospective participant in understanding the reasons why one might or might not want to participate in the research. This part of the informed consent must be organized and presented in a way that facilitates comprehension. The following language can be used for the first page of the consent form:
Research Study Summary:
- We are asking you to join a research study.
- The purpose of this research study is to [Insert the purpose of the study.]
- Study procedures will include: [Insert summary of study procedures.]
- [Number of visits] visits are required.
- These visits will take [Number of hours] hours total.
- There are some risks from participating in this study. [Insert summary of the study risks.]
- The study may have no benefits to you. [Describe benefits of the study to others. If there are expected benefits to subjects, revise the sentence and describe the benefits.]
- There are other choices available to you outside of this research. [Describe alternative procedures or treatments outside of the study. If none, you can delete the entire bullet point.]
- Taking part in this study is your choice. You can choose to take part, or you can choose not to take part in this study. You can also change your mind at any time. Whatever choice you make, you will not lose access to your medical care or give up any legal rights or benefits.
- If you are interested in learning more about the study, please continue reading, or have someone read to you, the rest of this document. Take as much time as you need before you make your decision. Ask the study staff questions about anything you do not understand. Once you understand the study, we will ask you if you wish to participate; if so, you will have to sign this form.
2. Include one of the two sentences regarding use of private information or identifiable biospecimens for future research (required):
- We will also share information about you with other researchers for future studies, but we will not use your name or any other identifiers. We will not ask you for any additional permissions.
OR
- We will not share any of your information with other researchers for future research studies, even if we remove all identifiers such as your name.
The next three elements are only required if they apply to your research. Review them and decide whether they also need to be included:
1. If you are collecting biospecimens: Add a statement that the participant’s biospecimens (even if identifiers are removed) may be used for commercial profit and that there are no plans to share the financial gains with him/her (unless there is a plan, then describe it).
2. If you are collecting biospecimens, explain whether the research will (if known) or might include whole genome sequencing (i.e., sequencing of a human germline or somatic specimen with the intent to generate the genome or exome sequence of that specimen) in the future.
3. If your research is likely to generate clinically relevant results: add a statement regarding whether clinically relevant research results, including individual research results, will be disclosed to the participant, and if so, under what conditions.
Important: If your study is sponsored by a sponsor external to Yale, please verify with the sponsor or the coordinating center that the research can be converted to the Revised Common Rule. Some of the sponsors specifically request that the research continues to be approved under Pre-Revised Common Rule.
The IRB will generally not require reconsent when incorporating these updates into the informed consent documents unless other significant changes are made. Once approved, the approval letter will list your obligations regarding submissions of modifications, reportable new information, and study closures.
Please, contact the HRPP Office at hrpp@yale.edu with any questions about this notification.
Sincerely,
Human Research Protection Program
Yale HRPP Plan to Implement Revised Regulation
January 9, 2019
The purpose of this announcement is to provide you with the Human Research Protection Program (HRPP) plans to implement the revised regulations governing research involving human subjects, the Common Rule. We refer to the revised regulations as the revised common rule or 2018 regulations throughout this notice.
Summary
The revised common rule was initially issued on January 19, 2017 and was to become effective on January 19, 2018. The implementation was delayed twice in the last year. The final implementation and compliance date is now January 21, 2019.
The revised rule is not applicable to FDA and DOJ (Department of Justice) regulated research. However, FDA plans to post a notice of draft revised FDA regulations intended to harmonize with the revised rule requirements. If you are involved in the conduct of FDA-regulated research, you should be aware that these changes may be directly applicable to your research in the future. We will let you know if and when those changes occur.
This communication addresses the following aspects of the revised common rule that may be important to you:
- Application of the revised common rule to research approved by the IRB prior to the implementation date–Grandfathered Research
- Minimal risk research
- Exempt research/modified and new exemption categories
- Informed consent requirements
- Single IRB of Record requirements
- IRES IRB system updates
- Changes in IRB practices to accommodate the revised rule
- Next Steps
1) Application of the revised common rule to research approved prior to the implementation date/Grandfathered Research
Research projects conducted at Yale University are held to varying standards depending upon what set of regulatory criteria applies to the research activity. As noted above, the revised rule is not applicable to FDA and DOJ (Department of Justice) regulated research.
Applicable research approved prior to January 21, 2019 will continue to be reviewed under the pre-2018 common rule regulations. Later this year, we will evaluate whether to transition applicable research approved prior to January 21, 2019 to the revised common rule at the time of continuing review. A communication about the plan will be sent out to the research community at that time.
2) Minimal Risk Research
The revised common rule allows for most minimal risk research to be conducted without IRB continuing review.
- The Yale IRB will NOT require continuing review for applicable minimal risk research approved after January 21, 2019.
- The Yale IRB will NOT require continuing review for greater than minimal risk research approved after January 21, 2019 if the research is closed to enrollment and the remaining research activities are limited to data analysis, including analysis of identifiable private information or identifiable specimens, or accessing follow-up clinical data from procedures that participants would undergo as part of clinical care.
- The Yale IRB will require continuing review for greater than minimal risk research (unless it meets criteria described above) and any research that is under FDA or DOJ jurisdiction regardless of the phase of the study.
Regardless of the continuing review category above, you must still report modifications and reportable new information to the IRB, including, but not limited to, changes in research or study personnel, study closures, events that may constitute serious or continuing noncompliance, and potential unanticipated problems involving risks to subject or others.
The Yale HRPP Office also will check in with the investigator on an annual basis regarding the progress of the study.
3) Exempt Research
The revised common rule changed the categories of research that can be deemed exempt.
- The revised exemption request form will be posted in the Library section of IRES IRB the week of January 14th.
- Two of the revised exemption categories include the use of broad consent (categories 7 & 8).
Due to lack of agency guidance related to requirements and use of broad consent, Yale IRB will generally not grant exemption determinations utilizing these categories. The Yale HRPP will re-evaluate the use of broad consent after the Department of Health and Human Services (HHS), Office for Human Research Protections (OHRP) issues relevant anticipated guidance. Due to the lack of guidance and large impact broad consent will have on institutions, the strategy to delay the use of broad consent is being taken by a clear majority of IRBs and academic institutions.
- The Yale IRBs will no longer issue exemptions under Yale-specific category 7. This category did not exist in the pre-2018 regulations and was created specifically for use by the Yale IRB and Yale researchers. The revised rule includes a new exemption category 3 (i) (A), (B), and (C), which allows for certain benign behavioral interventions to be considered exempt. As such, there is no further need for the largely redundant Yale-specific category 7.
Regardless of the exemption category, the requests for exemption determination must be submitted to the Yale IRB via IRES IRB per institutional requirements. Exempt research cannot commence without a Yale IRB exemption determination.
4) Informed Consent Requirements
Yale IRB will apply additional revised common rule consent provisions to new research approved by the IRB after January 21, 2019. Yale IRB will not apply the additional consent provisions of the revised common rule to research that is under FDA or DOJ jurisdiction unless it is also federally funded as it is not required for this research. New consent templates that reflect the new provisions will be posted in the Library section of IRES IRB. This includes a “concise and focused presentation of the key information” as required under the revised section .116 of the revised common rule, along with other changes. If your new study has been submitted and is under review by the Yale IRB on January 21, 2019 but is not yet approved, and the consent form submitted does not comply with the new regulatory requirements, a HRPP Regulatory Analyst will work with you to ensure that the new elements are added.
- Short Form: The IRB must still approve the use of a short form in research conducted at Yale. Under the revised rule the short form written informed consent document must now also state that the study key information was presented to the participant first before other information, if any, was provided. The translated versions of the short forms that are currently posted in the Library section of IRES IRB do not include the new required element. As such, the currently posted short forms can be used ONLY for studies that are not subject to the revised rule, such as research only under FDA or DOJ oversight (and without NIH, NSF, or other Common Rule entities’ funding), or research approved by the IRB prior to January 21, 2019. Revised short form templates for research subject to the revised rule will be available in IRES IRB in late January. Please, refer to 200 Guidance 2 on enrollment of non-English speaking individuals for instructions regarding how the short form must be used and documented.
5) Single IRB of Record
The revised rule requires a single IRB to review certain NIH-funded multi-site research. The Yale IRB generally does not serve as the single IRB of Record for multi-site research. We are working with external IRBs that can provide this service.
If you are planning a budget for a grant application for a multi-site research study, ensure that the budget includes appropriate provisions for the external IRB review fees. The Yale Center for Clinical Investigation offers a Research Budget Development service and can help investigators plan accordingly. The HRPP office can also assist. Please contact hrpp@yale.edu if you have questions regarding the external IRB budgeting process. For more information regarding the Yale HRPP Single IRB position, see also the Single IRB (sIRB) Review for Multi-site Research section on the News & Announcements page.
6) IRES IRB System Updates
We are working with a vendor and IRES IRB Support group to implement minor custom changes to the IRES IRB system that will help track studies approved under the revised rule. These changes will be implemented over the course of the next two weeks. A separate communication will be sent out when the change is made to the system.
7) Changes in IRB practices to accommodate the revised rule
Changes to the Approval Watermarks on Consent Forms and Protocol: Beginning January 21, 2019, Yale IRB-approved consent forms and research protocols will include an approval date only in the header and footer of the document. The approval watermark will no longer include a protocol expiration date. This change is necessary in order to manage the different regulatory requirements related to continuing review, and to allow for research no longer requiring continuing review per revised regulations to not have a IRES-IRB system-generated expiration date added to the consent form. Below is the explanation of the new practice for approvals of initial, modification, and continuing review submissions.
- New Research Study: The Yale IRBs will issue the consent and protocol documents with the initial approval date.
- Modification to the consent document: The Yale IRBs will issue the revised consent and protocol documents with approval date of the modification.
- Continuing Review: The Yale IRBs will issue the consent and protocol documents with the approval date of the continuing review.
The approval watermark on currently approved documents will not be changed until the next action on the protocol.
The principal investigator and the research study team is responsible for monitoring the expiration date of the study for continuing review purposes. To help with this responsibility, IRES IRB sends system-generated reminders to the PI and the PI Proxy 75 days, 45 days, and 30 days prior to the expiration date of the protocol. In addition, an expiring study report in IRES IRB is available to the investigator and study team at any time. The report includes a list of the studies associated with the PI in order of study expiration.
We recognize that this change may require adjustments to your practices regarding versioning of documents. For ideas regarding how to best track versions of your documents and keep track of the expiration dates, please, see Researcher Guide: Guide: Consent Version Control and Tracking located in the Help Center of IRES IRB.
Please note that the Yale HRPP may require specific versioning of documents in the future. When this occurs, we will inform you prior to the implementation of this change.
8) Next Steps
The Yale HRPP and Yale Center for Clinical Investigation (YCCI) will offer live training sessions that will focus on the changes to the regulations and how they affect research. We will also provide specialized training, including training for central campus. A separate communication will be sent when registration for the training offerings is available.
If your study is authorized for review by any of the external IRBs, the HRPP will work with the IRB to ensure the same implementation standard.
We value your opinions. If you have any questions or concerns regarding the implementation of the revised common rule, please send them to hrpp@yale.edu. We expect a large volume of inquiries and will compile them into an FAQ document for the research community and address them in the subsequent communication.
Thank you for your patience during this transition period.
Good Clinical Practice Training Announcement
November 6, 2018
Yale Human Research Protection Program and Yale Center for Clinical Investigation are hosting three in-person Good Clinical Practice training sessions available to the research community in November.
Initial Training: Essentials of Good Clinical Practice, Friday, November 30th from 9a.m. till 4p.m..
Refresher Training: Advanced Good Clinical Practice Training, Wednesday, November 28th, 5p.m. till 8p.m. OR Thursday, November 29th, 9a.m. till 12p.m..
The training will be held at Park St. Auditorium, #201 at 55 Park Street, New Haven. Lunch and/or refreshments will be provided.
Registration is required. Please, click on the name of the session and register through Training Management System (TMS). If you are unable to complete the registration using TMS, please contact irb.training@yale.edu.
Who needs to complete Good Clinical Practice training?
Good Clinical Practice training is required for anyone listed on a research study that meets the NIH definition of a clinical trial: A research study in which one or more human subjects are prospectively assigned to one or more interventions (which may include placebo or other control) to evaluate the effects of those interventions on health-related biomedical or behavioral outcomes.
How is a clinical trial indicated in IRES IRB?
Basic Information page of the IRES IRB application asks about prospective assignment of subjects and the existence of health-related or behavioral outcomes. If the answer to both questions is YES, the study is considered a clinical trial. A Good Clinical Practice requirement will be assigned in Training Management System the day after an individual is listed on that study.
How long is Good Clinical Practice training valid for?
The training is valid for 3 years. Refresher courses will satisfy the continuing education requirement.
Contact
You can email irb.training@yale.edu with any questions about the GCP and Human Subject Protection training requirements.
National Institutes of Health discontinuing the Human Subject Protection Training tutorial
September 14, 2018
On September 7, 2018 the National Institutes of Health announced their decision to cease the operation of their online tutorial ‘Protecting Human Research Participants’ as of September 26, 2018. The site will be shut down in its’ entirety, and users will not be able to access course certificates after that date. Archived course content will be available, but certificates will not. Individuals who require human subject protection training should access the Collaborative Intuitional Training Initiative (CITI) Program, available through the Yale Training Management Website.
Letter Templates Updated
September 06, 2018
In an effort to increase efficiency and readability, the HRPP revised format of the letters issued via the IRES IRB system, effective September 6, 2018. The new format more clearly delineates the type of review performed, streamlines references to documents reviewed, includes the Yale letterhead, as well as information that may be helpful for industry sponsors and other regulatory bodies.
Six Month Delay of the General Compliance Date of Revisions to the Common Rule
June 29, 2018
It was previously communicated that changes would be made to the Yale University HRPP and IRB review processes on July 19, 2018 in conformance with the effective date of the revised Common Rule. The Common Rule is a set of federal regulations governing a significant amount of human subjects research conducted at Yale University. Official notification has been published in the Federal Register that the Common Rule revisions have been further delayed with a new implementation date of January 21, 2019 (read more about the Federal Policy for the Protection of Human Subjects). During the delay, institutions may implement a limited number of the revised Common Rule “burden-reducing” provisions prior to the new implementation date. The Yale HRPP is in the process of reviewing these provisions and the feasibility of their implementation, which remain voluntary prior to January 21, 2019. Information on implementation of the revised Common Rule both before and after January 21, 2019 will be forthcoming.
European Union General Data Protection Regulation
May 25, 2018
Effective May 25, 2018, the European Union (EU) General Data Protection Regulation (GDPR) (EU 2016/679) replaces the 1995 Data Protection Directive (Directive 95/46/EC). The primary objective of this new regulation is to protect the fundamental rights and freedoms of data subjects and standardize requirements regarding covered data processing activities. Although the GDPR makes many important changes to EU data protection law, the GDPR is not a complete departure from existing principles. Many of the obligations outlined in the GDPR are not new for many organizations in the area of human subjects research. However, as noted below, the territorial reach of the new regulation has expanded and will impact U.S. organizations, including universities and academic medical centers.
The following Frequently Asked Questions (FAQs) provide an overview of the new regulation in the context of Human Subjects Research where the research involves the processing of personal data and special categories of personal data such as sensitive data or data concerning health. This document also briefly touches upon other key University activities that should be monitored for GDPR compliance.
View Frequently Asked Questions, European Union General Data Protection Regulation
Download Frequently Asked Questions, European Union General Data Protection Regulation
Single IRB (sIRB) Review for Multi-site Research
January 24, 2018
Overview of the NIH sIRB Policy
For applications with due dates on or after January 25, 2018, and contract solicitations published on or after January 25, 2018, NIH expects that all sites participating in multi-site studies, which involve non-exempt human subjects research funded by the NIH, will use a single Institutional Review Board (sIRB) to conduct the ethical review required for the protection of human subjects.
A sIRB model allows multiple institutions that are conducting the same protocol to use a single IRB for review, instead of using multiple IRBs to review the research at the sites individually.
The NIH policy applies to all biomedical and behavioral studies that:
- Are funded through grants, cooperative agreements, or contracts submitted to NIH on or after January 25, 2018, and
- Involve non-exempt human subjects research, and
- Involve multiple domestic sites, all of which are conducting the same protocol.
The sIRB NIH policy does not apply to:
- Foreign sites; or
- Studies funded through career development (K), institutional training (T), and fellowship (F) awards; or
- Sites for which federal, state, or tribal laws, regulations or policies require local IRB review; or
- collaborative projects in which multiple sites are involved but different sites may complete different parts of the study.
Other exceptions to allow for local IRB review may be considered by NIH based on compelling justification. These other exceptions must be reviewed and approved by NIH.
Click here for or more information regarding exceptions to the NIH sIRB Policy.
Single IRB Plan for Applications and Proposals
Applicants are expected to include a plan for the use of a sIRB in the grant applications and contract proposals submitted to the NIH (for due dates on or after January 25, 2018). For details, see Section 3.2 of the PHS Human Subjects and Clinical Trials Form Information Application Guide.
In general, your plan for the use of a sIRB should include a description regarding how you intend to comply with the sIRB policy and the name of the IRB that will serve as the IRB of record. For delayed multi-site research (studies in which there is no well-defined, detailed plan for human subjects involvement at the time of submission), the delayed onset justification attachment must include information regarding how the study will comply with the sIRB policy and state that a sIRB plan will be provided prior to initiating the study.
Designating the Single IRB in Applications and Proposals
For a NIH-funded multi-site project, investigators must designate the sIRB as part of the grant application and contract proposal. For example:
- The IRB at the lead PI’s site;
- The IRB at a participating site;
- The IRB at a non-participating site;
- Commercial IRBs (e.g., WCG/WIRB, Quorum Review, Advarra (previously Schulman and Chesapeake), Brany, and Hummingbird);
- An IRB specifically set up for an already-established-and-funded research network or consortium. This option is available only if the study you are proposing will be conducted under the auspices of the network/consortium.
- A Trial Innovation Network (TIN) IRB.
Note: In most situations, the lead PI, in collaboration with the HRPP at the lead PI’s institution, will select the sIRB. The selected IRB must be willing to serve as the sIRB and all of the participating sites must agree to rely on the sIRB. The IRB also must be registered with the Office for Human Research Protections (OHRP) to serve as a sIRB.
Yale approved IRBs for sIRB
When a Yale Investigator is the awardee of a multi-site grant, Yale plans to cede IRB oversight to an external IRB to serve as the sIRB on Yale’s behalf. If a commercial IRB is selected, Yale will cede IRB oversight to one of the 5 external IRBs below.
- Advarra (previously known as Schulman and Chesapeake)
- Brany
- Hummingbird
- Quorum Review
- WCG/WIRB
If you have questions regarding any of the commercial IRBs, please contact the HRPP office at hrrp@yale.edu.
If you prefer to employ services of a commercial IRB not listed above, please contact the HRPP office at hrpp@yale.edu prior to submitting the grant application as there may be additional contractual considerations that must be taken into account when choosing an external IRB that does not have a master agreement with Yale.
If there is a compelling reason for the Yale IRB to serve as the sIRB for a specific study, the HRPP will evaluate such a request on a case-by-case basis.
Cost of Single IRB Review - sIRB Budget in the Grant Application
The costs for IRB review at a single institution by that institution’s IRB have typically been considered an indirect cost covered under an institution’s Facilities and Administration (F&A) rate (except for industry-initiated-and-sponsored studies). NIH, however, expects that many sIRBs will charge fees to serve as a single IRB and review other sites. The grant budget must therefore include appropriate provisions for IRB review fees to avoid gaps in coverage.
The Yale Center for Clinical Investigation (YCCI) offers a Research Budget Development service and can help investigators with budgeting. Please contact clinicalresearchresources@yale.edu if you have questions. You may also contact the HRPP office at hrpp@yale.edu if you have questions or need information regarding commercial IRB pricing for any of the 5 IRBs below.
- Advarra (previously known as Schulman and Chesapeake)
- Brany
- Hummingbird
- Quorum Review
- WCG/WIRB
Request for HRPP approval to use an External IRB
A request to use an external IRB for the review of research conducted by a Yale investigator must be submitted to the HRPP Office through IRES IRB. A submission to an external IRB may not be made without prior authorization from the Yale HRPP.
Although agreements with external IRBs may include different division of responsibilities, Yale as an institution will generally remain responsible for review of compliance with local requirements such as training, locally required ancillary reviews, conflict of interest disclosures, or Yale specific language in the consent forms.
Letter of Support
If you are requested to provide an institutional letter of support to cede review to a specific sIRB for your grant application, please contact the HRPP office at hrpp@yale.edu. In your request, please include the name of the PI at the Yale site, the name of the proposed single IRB, the RFA number, and the funding opportunity title.
Billing Contact for External sIRB Review
When Yale cedes review to an external IRB, Yale HRPP is listed as the primary billing contact and YCCI (or the PI’s business office or other designated billing department) as the secondary contact. The HRPP will work with the external IRB and designated billing contact to ensure payment to the external IRB.
Primary Billing Contact
Human Research Protection Program (HRPP)
hrpp@yale.edu
Attention: Meriam Worzella
Secondary Billing Contact
Yale Center for Clinical Investigation (YCCI)
Research Budget Development Unit
clinicalresearchresources@yale.edu
Attention: Juliann Murphy
Note: If YCCI is not the correct secondary billing contact, the Research Budget Development Unit will direct you to correct business office contact for the specific study.
Questions
If you have questions about the NIH policy or questions about choosing a single IRB for your upcoming grant applications, you may contact hrpp@yale.edu. Additional guidance is forthcoming. Training sessions regarding the use of external IRBs and reliance agreements will be offered soon.
NIH Resources
- NOT-OD-16-094 Final NIH Policy on the Use of a Single Institutional Review Board for Multi-Site Research
- NOT-OD-17-076 Revision: Notice of Extension of Effective Date for Final NIH Policy on the Use of a Single Institutional Review Board for Multi-Site Research
- NOT-OD-16-109 Scenarios to Illustrate the Use of Direct and Indirect Costs for Single IRB Review under the NIH Policy on the Use of a Single IRB for Multi-site Research
- NOT-OD-18-003 Guidance on Exceptions to the NIH Single IRB Policy
- NOT-OD-18-004 Guidance on Implementation of the NIH Policy on the Use of a Single Institutional Review Board for Multi-Site Research
- Frequently Asked Questions
Delay in the Implementation of the Revised Common Rule
January 17, 2018
This is an important update regarding revisions to the regulations governing research involving human subjects, the Common Rule. The revised rule was issued on January 19, 2017 and it was to be become effective on January 19, 2018. The revised rule was not applicable to FDA and DOJ (Department of Justice) regulated research.
On January 17, 2018, the Department of Health and Human Services issued a statement that the effective and compliance date of the new rule is delayed until July 19, 2018.
Over the past few months, Yale HRPP has worked on implementation of the revised regulations. This included updates to policies and procedures, submission forms, consent templates, and IRES IRB system configuration. The delay means that we cannot implement these changes until July 19, 2018, unless there is a decision to further delay the revised Common Rule beyond the July 19, 2018 date. This additional time will allow the regulatory agencies to provide guidance necessary for institutions to implement the rule. It is also hoped that FDA will have issued revised regulations intended to harmonize with the new requirements by that time. This would eliminate confusion that may result from applying different regulatory standards.
You may have read about the changes and wondered how they will affect your research. Yale HRPP and YCCI will offer live training sessions in the next few months that will focus on the proposed changes to the regulations. You can sign up for Coffee and Conversation and Lunch and Learn series at yale.edu/training.
We value your opinions. If you have any questions or concerns regarding this notification, please send them to hrpp@yale.edu. We expect a large volume of inquiries and will compile them into a FAQ document for the research community and address them in a subsequent communication.
Thank you for your patience during this transition period.
January 8, 2018
Effective January 2, 2018: As a result of a joint effort to streamline the submission process regarding studies involving minors that require approval from the Pediatric Protocol Review Committee (PPRC), research protocols can now be submitted in IRES IRB without first obtaining PPRC approval outside of the system.
When a study is submitted into IRES IRB, the HRPP Office will request PPRC review using the functionality available in the IRES IRB system. The HRPP pre-review comments along with any PPRC required revisions will be provided to the PI and the PI Proxy via the IRES IRB system. Once the PPRC approval is obtained, the research protocol will be scheduled to the IRB for review and approval.
We hope this change will improve your experience with submissions of protocols involving minors.
Effective January 18, 2018: Biomedical studies will be assigned to the next available weekly IRB meeting regardless of the department the study comes from. Oncology studies will continue to be scheduled to the oncology-focused IRB meetings on the 1st and 3rd week of the month, but they can also be sent to a week 2 or 4 biomedical IRB panel. The purpose of the change is to improve the Yale internal IRB’s timelines for review and approval.
Designated HRPP staff will continue to work with investigators to ensure that the submissions are complete and ready for review by the IRB prior to scheduling to a meeting. To accommodate this change, an additional monthly meeting will be added to the first week of every month. Updated IRB Rosters and an IRB meeting schedule will be available and posted on the HRPP website in early January.
We are also happy to welcome new members to our HRPP family. Jessica (Jessie) Huening joined the office as Assistant Director, Regulatory, Compliance & Quality in early December. Dianna Mercier and Griselle Gandulla joined the office as IRB Regulatory Analysts in October 2017.
Most importantly, we thank our dear colleague and mentor, Dr. Sandra Alfano, for her many years of service to the HRPP and HIC. We will greatly miss her guidance and presence in the HRPP Office and wish her well in her new role as Senior Research Analyst with the Yale Conflict of Interest Committee.
Please, see below for the revised assignment of the Chairs and the IRB panels.
Week 1:
- HIC IA: Wednesday, 3:00 PM (added meeting)
- Chair: Madelon Baranoski, Ph.D.
- Vice-Chair: Stephen Latham, JD, PhD
- HIC IB: Wednesday, 5:00 PM
- Chair: John Roberts, MD
- Vice Chair: Madelon Baranoski, Ph.D.
Week 2:
- HIC II: Wednesday, 5:00 PM
- Chair: Suchitra Krishnan-Sarin, PhD
- Vice-Chair: Stephen Latham, JD, PhD
Week 3:
- HIC III: Wednesday, 3:00PM
- Chair: Madelon Baranoski, Ph.D.
- Vice-Chair: Suchitra Krishnan-Sarin, Ph.D.
- HIC IB: Friday, 7:30AM
- Chair: John Roberts, MD
- Vice-Chair: Madelon Baranoski, Ph.D.
Week 4:
- HIC IV: Wednesday, 5:00PM
- Chair: Suchitra Krishnan-Sarin, PhD
- Vice-Chair: Stephen Latham, JD, PhD
Should you have questions about the full board reviews and schedule, please contact any of the designated Full Board IRB Regulatory Analysts: Dawn Pedevillano, Stacey Scirocco, Theresa Katz, Dianna Mercier, or Griselle Gandulla.
The Human Research Protection Program (HRPP) staff wish you a very Happy New Year 2018!
Dear Research Community,
We have a few announcements to make before the winter recess begins on December 22.
Effective January 2, 2018: As a result of a joint effort to streamline the submission process regarding studies involving minors that require approval from the Pediatric Protocol Review Committee (PPRC), research protocols can now be submitted in IRES IRB without first obtaining PPRC approval outside of the system.
When a study is submitted into IRES IRB, the HRPP Office will request PPRC review using the functionality available in the IRES IRB system. The HRPP pre-review comments along with any PPRC required revisions will be provided to the PI and the PI Proxy via the IRES IRB system. Once the PPRC approval is obtained, the research protocol will be scheduled to the IRB for review and approval.
We hope this change will improve your experience with submissions of protocols involving minors.
Effective January 18, 2018: Biomedical studies will be assigned to the next available weekly IRB meeting regardless of the department the study comes from. Oncology studies will continue to be scheduled to the oncology-focused IRB meetings on the 1st and 3rd week of the month, but they can also be sent to a week 2 or 4 biomedical IRB panel. The purpose of the change is to improve the Yale internal IRB’s timelines for review and approval.
Designated HRPP staff will continue to work with investigators to ensure that the submissions are complete and ready for review by the IRB prior to scheduling to a meeting. To accommodate this change, an additional monthly meeting will be added to the first week of every month. Updated IRB Rosters and an IRB meeting schedule will be available and posted on the HRPP website in early January.
We are also happy to welcome new members to our HRPP family. Jessica (Jesse) Huening joined the office as Assistant Director, Regulatory, Compliance & Quality earlier this month. Dianna Mercier and Griselle Gandulla joined the office as IRB Regulatory Analysts in October 2017.
Most importantly, we thank our dear colleague and mentor, Dr. Sandra Alfano, for her many years of service to the HRPP and HIC. We will greatly miss her guidance and presence in the HRPP Office and wish her well in her new role as Senior Research Analyst with the Yale Conflict of Interest Committee.
The HRPP Office will be closed for winter recess from December 22, 2017 until January 2, 2018. During this time, any urgent correspondence and issues related to subject safety should be directed to the HRPP Director, Linda Colman, at linda.coleman@yale.edu. We will also periodically monitor the correspondence sent to hrpp@yale.edu, but will not be able to process any submissions until we return in January.
The Human Research Protection Program (HRPP) staff wish you a relaxing winter holiday and a Happy New Year!
Notice of Changes to NIH Policy for Issuing Certificates of Confidentiality
Effective October 1, 2017, all NIH-funded research that was commenced or ongoing on or after December 13, 2016 and collects or uses identifiable, sensitive information is deemed to be issued a Certificate of Confidentiality, and is therefore required to protect the privacy of individuals who are subjects of such research in accordance with subsection 301(d) of the Public Health Service Act.
Note:
- “Identifiable, sensitive information” means information about an individual that is gathered or used during the course of biomedical, behavioral, clinical, or other research, where the following may occur:
- An individual is identified; or
- For which there is at least a very small risk, that some combination of the information, a request for the information, and other available data sources could be used to deduce the identity of an individual.
- The NIH will continue to consider request for Certificates for non-federally funded research in which identifiable, sensitive information is collected or used.
- For more information, please visit the Notice of Changes to NIH Policy for Issuing Certificates of Confidentiality website
- Visit the National Institutes of Health’s internet site on Certificates of Confidentiality website
- Visit CoC Contacts at NIH and Other DHHS Agencies that Issue Certificate website
- Please contact the Yale HRPP Office (hrpp@yale.edu) with any questions. Training materials on CoCs are being developed.
April 28, 2017
The HRPP office has received questions regarding ClinicalTrial.gov disclosure requirements. To assist in better understanding of the ClinicalTrials.gov registration and reporting requirements, please refer to HRPP Policy 1000, Clinical Trial Registration and Results Reporting Requirement. See also the 04/27/2017 Office of Research Administration notice.
Principal Investigators may contact the Yale Center for Clinical Investigation (YCCI) for assistance with ClinicalTrials.gov registration and reporting for more information.
Principal Investigators who are unsure as to whether a clinical trial is subject to ClinicalTrial.gov disclosure requirements should contact Yale.CTgov@yale.edu or HRPP@yale.edu for assistance.
Monday, April 3, 2017
A revised HRPP procedure 920 PR.4 titled ‘Use of External IRBs for Review and Oversight of Research Involving Human Subjects’ went into effect Monday, April 3, 2017.
The revised procedure allows the HRPP to consider and allow the use of external (commercial) IRBs for a broader range of research studies. The procedure delineates the process in which Yale investigators can request review by an external IRB and describes the responsibilities of Yale, the investigator, and the external IRB after the study is approved for external IRB review.
Currently, Yale maintains agreements with Western IRB (WIRB) and Quorum Review. Agreements with additional IRBs including BRANY, Chesapeake, Hummingbird, and Schulman will be finalized soon. Each of the IRBs will be invited to provide live training to the Yale research community regarding the use of their electronic portals, protocol submission processes, reporting requirements, and other aspects of their operations and review capabilities. Announcements with training dates will be emailed later this month.
A quick guide on requesting review by an external IRB is available in the Help Center section in IRES IRB. Questions regarding the revised procedure can be sent to HRPP@yale.edu.
Good Clinical Practice Training Requirement
January 2, 2017
The requirement for Good Clinical Practice training for all investigators and staff involved in the conduct, oversight, or management of clinical trials was announced to the research community on December 22, 2016. The details regarding the training are available in the memo from Pamela Caudill, Senior Associate Provost for Research Administration.
New IRES IRB Electronic Protocol Submission System
January 2, 2017
The HRPP is pleased to announce a new IRES IRB electronic protocol submission system is LIVE for our research community!
The first group of researchers to migrate over to the new system was in July 2016. Other departments migrated over in the weeks/months after with all departments migrated in December 2016. Training sessions have been offered prior to each department going live.
Many hours have been dedicated to making this new system user friendly, reliable and efficient. We strived to provide Principal Investigators and study personnel with a system that aids in a smoother protocol submission, review and approval process.
Thank you for working with us throughout this exciting transition period! Any questions please contact IRB.Support@yale.edu.
Consent Consultations
August 26, 2016
The Yale IRB staff is available to meet with research coordinators, Principal Investigators, and consenting personnel to help create a consent process checklist and practice obtaining consent from subjects using a study approved consent form. Individual appointments will be held in the offices at 55 College Street and will last between 45 minutes and an hour. For more information and to schedule an appointment, contact Michele Antisdel at michele.antisdel@yale.edu or Jennifer Reese at jennifer.reese@yale.edu.
New IRES IRB Electronic Protocol Submission System
The HRPP is pleased to announce that a new IRES IRB electronic protocol submission system for our research community will begin rolling out as of July 12, 2016.
The first group of researchers to migrate over to the new system will be the Cardiology Department, whose protocols are reviewed by the HIC III committee. Once this group is fully functioning with the system, we will begin migrating other departments. We will ensure that all departments are given advance notice of their migration, and we will schedule training sessions prior to each department going live.
Many hours have been dedicated to making this new system user friendly, reliable and efficient. We strived to provide PIs and study personnel with a system that aids in a smoother protocol submission, review and approval process.
Departments who have not yet been notified that they are going live with IRES IRB will continue to submit as usual (e.g., if you use Coeus, continue to use Coeus; if you email your submissions, continue to email). The IRB office will be using Coeus, email, and IRES IRB submission systems concurrently until everyone has been transitioned to using IRES IRB.
Thank you for working with us throughout this exciting transition period! Any questions please contact IRB.Support@yale.edu
Attention Research Community!
Hartford HealthCare (HHC) is hosting a national conference with OHRP that you might find interesting: “Foundations of Trust: Connecting Our Community to Research.”
- When: October 25-26, 2016
- Where: Hartford Marriott Downtown Hotel
See attached flyer for more information.
Policy, Procedure and Guidance - IMPORTANT UPDATES (December 18, 2015)
Policy 100: Revised criteria for granting Category 7 Exemption to apply only to unfunded research
Guidance 100 GD2: Revised criteria for granting 2-year extended approval period to apply only to unfunded research
Visit our //calendar.google.com/calendar/embed?src=monika.lau@yale.edu">HRPP Training Calendar.
RDRC Announcement
Newly formed Yale University Radioactive Drug Research Committee (Yale RDRC)
Yale University has established a new FDA approved Radioactive Drug Research Committee (RDRC) with Dr. Albert Sinusas as the committee Chair. Yale New Haven Hospital (YNHH) will be closing out the YNHH RDRC, and transferring all protocols to the Yale RDRC. Faculty researchers conducting research at the University that requires RDRC approval should no longer submit these applications to the Yale New Haven Hospital RDRC as this committee is not receiving or reviewing research protocols going forward. The Yale RDRC will continue to work in conjunction with the Yale University Radiation Safety Committee (RSC).
On behalf of the Yale University academic community we would like to acknowledge Dr. Ravinder Nath and the YNHH RDRC committee members for their tremendous efforts in operation of the RDRC on behalf of Yale University investigators for many years.
Yale University Radioactive Drug Research Committee (Yale RDRC):
The information and instructions on this website will be modified and updated with operation of the new Yale RDRC, so please check this website prior to any submissions for changes. View our policies and forms.
For questions regarding the application process, approvals and/or future committee meeting dates and deadlines, please contact Donna McMahon at RDRC_admin@yale.edu.
IRB Forms Notice
The Yale University Institutional Review Board (HIC/HSC) periodically revises forms that are required for submission (e.g., new application, renewal form, amendment form, etc.). Changes to the forms generally are based on information that is required to provide the reviewer with accurate and up-to-date information and/or due to changes in regulations or University policies. You must always use the form that is currently posted on the HRPP forms website. All forms are periodically reviewed to meet changing regulatory or other requirements. For this reason, applications submitted on forms that are outdated by more than 2 months will not be accepted. To ensure that you are using the most current form, please always refer to the HRPP website or the COEUS link each time you need to complete a form for your specific submission. DO NOT keep template forms on your computer.
Examples of requirements and questions:
- “Request for Approval of a Social/Behavioral/Educational Study”. This form, which is posted on both the social/behavioral and biomedical forms pages, provides a structured application format for those studies that do not include treatment interventions.
- List the Business Manager and contact information.
- Funding Source: Indicate ALL of the CURRENT and ACTIVE funding source(s) for this study.
- Has the funding source changed since this HIC application was last renewed?
- Did the protocol lapse?If, so complete the following questions (omitted here)
IRB Fees
Please note, Effective January 1, 2015, HIC protocols with start date on or after January 1, 2015, will be subject to the new IRB fees at prevailing rates. These rates are NOT locked for the duration of the contract. The change in IRB fees structure is based on a 2014 study of IRB fees charged for industry-sponsored applications across the United States. Visit the IRB Fees Schedule.
Reminder to Investigators Regarding NIH JIT Submissions
As of June 16, 2014, for NIH JIT Submissions, a protocol /proposal congruency form (100 FR26) completed by the proposal PI must be submitted to the HIC/HSC as part of the protocol application or amendment process. The form is posted on our website under Forms.
Important Update
Revised IRB Policy 710: REPORTING UNANTICIPATED PROBLEMS INVOLVING RISKS TO SUBJECTS OR OTHERS, INCLUDING ADVERSE EVENTS, TO THE IRB
Effective April 15, 2014, the Yale University Human Research Protection Program (HRPP) revised IRB Policy 710 and related procedures (the policy and procedure for reporting adverse events and unanticipated problems involving risks to subjects or others (UPIRSOs) to the IRB). The changes are as follows:
- The revised policy requires reporting of internal events (i.e., events that occur at a study site under the jurisdiction of a Yale IRB) within 5 calendar days of the Principal Investigator becoming aware of the event.
- The revised policy requires, to the extent possible, immediate notification to the IRB of all events that might require a temporary or permanent interruption of research activity, and
- The revised policy requires reporting of external events (i.e., events that occur at a study site NOT under the jurisdiction of a Yale IRB) within 15 calendar days of the Principal Investigator becoming aware, but only if an amendment to the protocol and/or consent documentation is necessary at Yale.
To aid in understanding reportable events, the new policy includes a decision tree for study personnel to use. In addition, a single reporting form has replaced the previous reporting forms and has been designed to capture required information, to direct the researcher in assessing the event, and to capture the IRB process of event review.
The revised Policy 710 and related documents went in effect as of April 15, 2014. Please review the revised documents carefully. If you have any questions, submit them via email to hrpp@yale.edu.